¶ Intrinsically Disordered Regions
Intrinsically disordered regions (IDRs), also known as intrinsically unstructured protein regions (IUPRs), intrinsically disordered protein regions (IDPRs) are protein regions which lack a stable tertiary (3D) structure. If the entire length of protein is unstructured then the protein is considered an intrinsically unstructured/disordered protein (IUP/IDP). The use of the 'region' nomenclature is used to refer specifically to the unstructured parts of the protein as opposed to its entirity.
¶ Definition of Disorder
The definition of protein disorder varies according to the biological field. A thermodynamics-based definition of disorder based on the native energy landscape of the protein identifies disordered regions where there are multiple local energy minima which can be traversed without high activation energy [1]; however the energy landscape of a particular protein is not easily comprehensively measured. A structural biology-based interpretation of disorder may revolve around the ability to solve for a 3D structure using crystallography or electron-microscopy, however this is limited in its throughput. High-throughput bioinformatic studies use biophysical energetic approximations (e.g. IUPRED [2]) or machine learning algorithms (e.g. DISOPRED)[3][4] on known disordered proteins to predict disorder in a protein sequence.
¶ Characteristics of IDRs
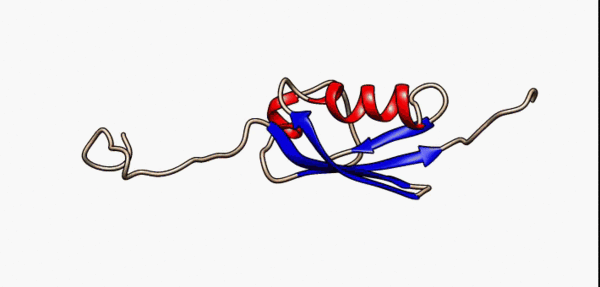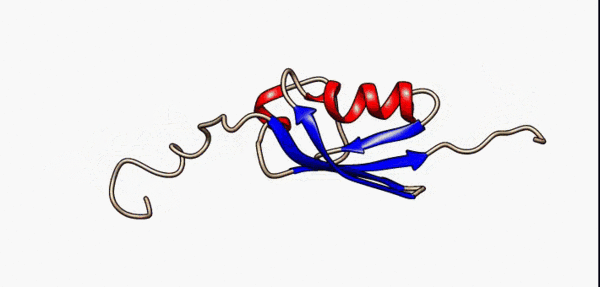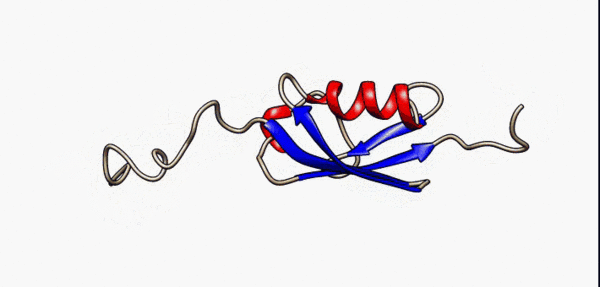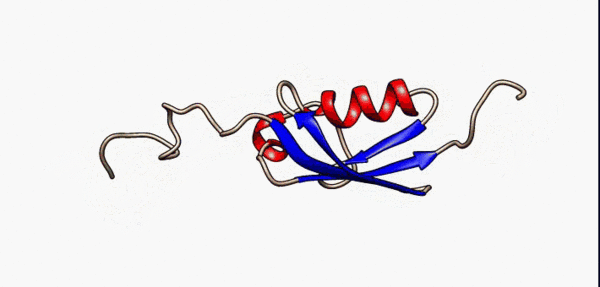
N- and C-terminal disordered regions (grey) of the SUMO-1 protein are highly flexible as modelled by nuclear magnetic resonance [5]
As a consequence of their lack of structure, IDRs are inherently flexible, particularly in comparison to structured/ordered domains, facilitating their binding capabilities [6]. In terms of sequence, IDRs also tend to be lower in complexity than ordered regions and tend to be more repetitive since they use a biased subset of the amino acid alphabet enriched in proline, arginine, glycine, glutamine, serine, lysine, alanine and glutamate[7][8][9][10][5:1]. IDRs also harbor a variety of interacting motifs including short linear interacting motifs (SLiMs) or molecular recognition features (MoRFs), further enhancing their binding capabilities by increasing valency[6:1]. As a result, IDRs are highly important promoting condensate formation, particularly as scaffolds.
Boehr, D. D., Nussinov, R. & Wright, P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nature Chemical Biology vol. 5 789–796 (2009). ↩︎
Mészáros, B., Erdös, G. & Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337 (2018). ↩︎
Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F. & Jones, D. T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 337, 635–645 (2004). ↩︎
Ward et al. (2004), The DISOPRED server for the prediction of protein disorder. ↩︎
Majorek K, Kozlowski L, Jakalski M, Bujnicki JM (December 18, 2008). "Chapter 2: First Steps of Protein Structure Prediction" (PDF). In Bujnicki J (ed.). Prediction of Protein Structures, Functions, and Interactions. John Wiley & Sons, Ltd. pp. 39–62. doi:10.1002/9780470741894.ch2. ISBN 9780470517673. ↩︎ ↩︎
van der Lee et al. (2014), Classification of Intrinsically Disordered Regions and Proteins. ↩︎ ↩︎
Romero, P. et al. Sequence complexity of disordered protein. Proteins Struct. Funct. Genet. 42, 38–48 (2001). ↩︎
Vacic, V., Uversky, V. N., Dunker, A. K. & Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics 8, (2007). ↩︎
Williams, R. M. et al. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 89–100 (2001). doi:10.1142/9789814447362_0010 ↩︎
Campen, A. et al. TOP-IDP-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder. Protein Pept. Lett. 15, 956–963 (2008). ↩︎